1
Introduction
1 min•156 words
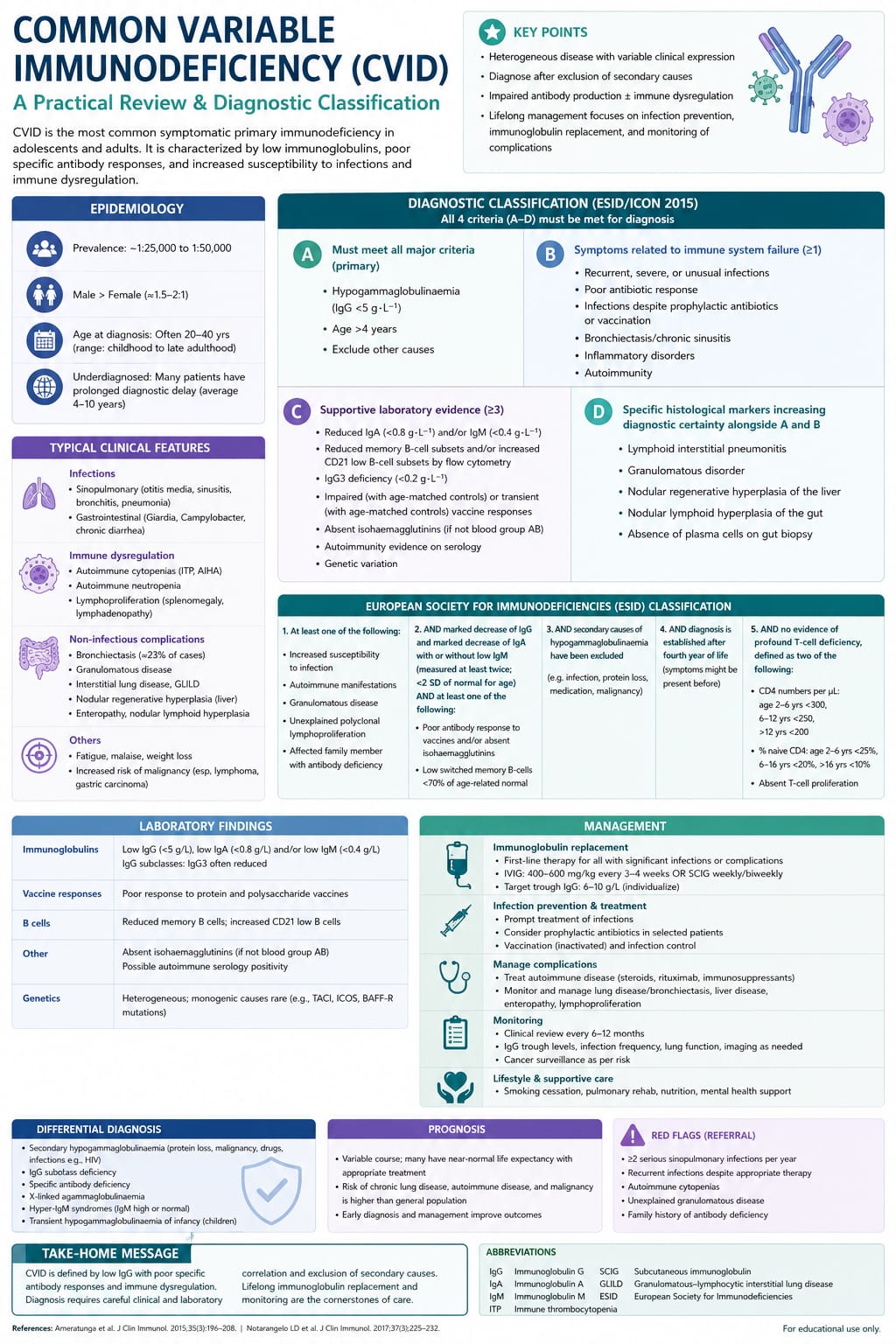
Common Variable Immunodeficiency (CVID) is the most prevalent symptomatic primary immunodeficiency disorder encountered in clinical practice. Characterized by a profound disruption in humoral immunity, CVID is defined by significantly reduced serum levels of IgG accompanied by low levels of IgA, IgM, or both, alongside a compromised or completely absent antibody response to vaccinations or natural infections.
While classified as a primary immunodeficiency, CVID is unique because its clinical onset is highly variable, most frequently presenting in late adolescence or early adulthood between the second and fourth decades of life. It is a profoundly heterogeneous disorder, and the "variable" in its name underscores not only the unpredictable timing of its clinical debut but also the highly diverse spectrum of manifestations it produces.
Beyond a baseline susceptibility to recurrent pyogenic infections, a massive subset of CVID patients suffer from severe, non-infectious complications driven by immune dysregulation, including autoimmunity, chronic granulomatous disease, enteropathy, and an elevated risk of malignancy.
While classified as a primary immunodeficiency, CVID is unique because its clinical onset is highly variable, most frequently presenting in late adolescence or early adulthood between the second and fourth decades of life. It is a profoundly heterogeneous disorder, and the "variable" in its name underscores not only the unpredictable timing of its clinical debut but also the highly diverse spectrum of manifestations it produces.
Beyond a baseline susceptibility to recurrent pyogenic infections, a massive subset of CVID patients suffer from severe, non-infectious complications driven by immune dysregulation, including autoimmunity, chronic granulomatous disease, enteropathy, and an elevated risk of malignancy.
2
Background & Pathophysiology
2 min•237 words
The underlying pathophysiology of Common Variable Immunodeficiency is centered on a failure of B-lymphocyte differentiation into mature, immunoglobulin-secreting plasma cells and memory B-cells.
In the vast majority of patients with CVID, the total number of circulating peripheral blood B-cells remains within normal limits. However, phenotypic tracking reveals a structural block in the late stages of B-cell maturation. Specifically, there is a severe reduction in switched memory B-cells, which are crucial for maintaining long-term humoral protection.
From a genetic standpoint, most cases of CVID are sporadic, but roughly ten to twenty percent of patients demonstrate a familial pattern of inheritance, typically autosomal dominant with variable penetrance. To date, specific monogenic mutations have been identified in less than twenty percent of patients. These mutations map to critical genes regulating B-cell survival, activation, and costimulation, including:
ICOS (Inducible T-cell Costimulator): Crucial for T-cell dependent B-cell activation and germinal center formation.
TNFRSF13B (Encoding TACI): Involved in isotype switching and plasma cell survival. Mutations in TACI are the most common genetic variants associated with CVID.
CD19, CD20, and CD81: Core component receptors that form the essential B-cell co-receptor complex required for signal transduction.
This breakdown in humoral signaling triggers a profound multi-system vulnerability. While the lack of protective antibodies permits recurrent bacterial invasions, the concurrent dysregulation of the cellular immune arm—manifesting as T-cell cytopenias, restricted T-cell receptor repertoires, and decreased regulatory T-cells—directly fuels a state of chronic, systemic autoinflammation and tissue destruction.
In the vast majority of patients with CVID, the total number of circulating peripheral blood B-cells remains within normal limits. However, phenotypic tracking reveals a structural block in the late stages of B-cell maturation. Specifically, there is a severe reduction in switched memory B-cells, which are crucial for maintaining long-term humoral protection.
From a genetic standpoint, most cases of CVID are sporadic, but roughly ten to twenty percent of patients demonstrate a familial pattern of inheritance, typically autosomal dominant with variable penetrance. To date, specific monogenic mutations have been identified in less than twenty percent of patients. These mutations map to critical genes regulating B-cell survival, activation, and costimulation, including:
ICOS (Inducible T-cell Costimulator): Crucial for T-cell dependent B-cell activation and germinal center formation.
TNFRSF13B (Encoding TACI): Involved in isotype switching and plasma cell survival. Mutations in TACI are the most common genetic variants associated with CVID.
CD19, CD20, and CD81: Core component receptors that form the essential B-cell co-receptor complex required for signal transduction.
This breakdown in humoral signaling triggers a profound multi-system vulnerability. While the lack of protective antibodies permits recurrent bacterial invasions, the concurrent dysregulation of the cellular immune arm—manifesting as T-cell cytopenias, restricted T-cell receptor repertoires, and decreased regulatory T-cells—directly fuels a state of chronic, systemic autoinflammation and tissue destruction.
3
Clinical Features
2 min•380 words
The clinical presentation of Common Variable Immunodeficiency is classically split into two primary phenotypes: the infectious phenotype, which is universal, and the dysregulatory or non-infectious phenotype, which affects roughly half of all diagnosed patients.
1. Recurrent Sino-Pulmonary Infections
The universal hallmark of CVID is a history of frequent, severe, or recurrent bacterial infections involving the respiratory tract. Patients present with repetitive episodes of acute otitis media, severe sinusitis, and recurrent bacterial pneumonia. The predominant pathogens isolated include encapsulated bacteria, specifically Streptococcus pneumoniae and Haemophilus influenzae.
Without early diagnosis and intervention, these recurrent lower respiratory infections cause progressive, irreversible damage to the pulmonary architecture, culminating in diffuse bronchiectasis, chronic obstructive lung disease, and secondary cor pulmonale.
2. Autoimmune Manifestations
Autoimmunity is the most common non-infectious complication of CVID, frequently serving as the initial presenting sign of the disease before sinus or lung infections emerge. Immune-mediated cytopenias are highly characteristic, specifically Immune Thrombocytopenia (ITP) and Autoimmune Hemolytic Anemia (AIHA). When these two cytopenias occur concurrently, it is known as Evans Syndrome and should immediately trigger an evaluation for an underlying immunodeficiency. Other associated autoimmune conditions include Rheumatoid Arthritis, Systemic Lupus Erythematosus, and vitiligo.
3. Gastrointestinal Enteropathy
CVID-associated enteropathy affects a significant portion of patients, manifesting as chronic, watery diarrhea, severe malabsorption, abdominal cramping, and progressive weight loss. Endoscopic biopsy reveals a picture that closely mimics Celiac Disease, showing villous atrophy and crypt hyperplasia, but with a pathognomonic absence of plasma cells in the lamina propria. This enteropathy can also be exacerbated by persistent opportunistic infections, particularly chronic Giardiasis or Salmonella infestations.
4. Granulomatous and Lymphoproliferative Disease
Immune dysregulation frequently induces non-caseating granuloma formation across multiple organs, highly resembling Sarcoidosis. These granulomas can infiltrate the lungs (known as Granulomatous-Lymphocytic Interstitial Lung Disease or GLILD), the liver (causing hepatomegaly and portal hypertension), and the spleen. Generalized lymphadenopathy and splenomegaly are present on physical examination in up to one-third of patients, reflecting benign but extensive lymphoid proliferation.
5. Increased Malignancy Risk
Patients with CVID carry a significantly elevated baseline risk for developing malignancies. There is a remarkably high incidence of gastric carcinoma, heavily associated with a higher rate of Helicobacter pylori infections and achlorhydria. Furthermore, the risk of developing non-Hodgkin lymphoma is increased up to 30-fold compared to the general population, typically emerging during adulthood.
1. Recurrent Sino-Pulmonary Infections
The universal hallmark of CVID is a history of frequent, severe, or recurrent bacterial infections involving the respiratory tract. Patients present with repetitive episodes of acute otitis media, severe sinusitis, and recurrent bacterial pneumonia. The predominant pathogens isolated include encapsulated bacteria, specifically Streptococcus pneumoniae and Haemophilus influenzae.
Without early diagnosis and intervention, these recurrent lower respiratory infections cause progressive, irreversible damage to the pulmonary architecture, culminating in diffuse bronchiectasis, chronic obstructive lung disease, and secondary cor pulmonale.
2. Autoimmune Manifestations
Autoimmunity is the most common non-infectious complication of CVID, frequently serving as the initial presenting sign of the disease before sinus or lung infections emerge. Immune-mediated cytopenias are highly characteristic, specifically Immune Thrombocytopenia (ITP) and Autoimmune Hemolytic Anemia (AIHA). When these two cytopenias occur concurrently, it is known as Evans Syndrome and should immediately trigger an evaluation for an underlying immunodeficiency. Other associated autoimmune conditions include Rheumatoid Arthritis, Systemic Lupus Erythematosus, and vitiligo.
3. Gastrointestinal Enteropathy
CVID-associated enteropathy affects a significant portion of patients, manifesting as chronic, watery diarrhea, severe malabsorption, abdominal cramping, and progressive weight loss. Endoscopic biopsy reveals a picture that closely mimics Celiac Disease, showing villous atrophy and crypt hyperplasia, but with a pathognomonic absence of plasma cells in the lamina propria. This enteropathy can also be exacerbated by persistent opportunistic infections, particularly chronic Giardiasis or Salmonella infestations.
4. Granulomatous and Lymphoproliferative Disease
Immune dysregulation frequently induces non-caseating granuloma formation across multiple organs, highly resembling Sarcoidosis. These granulomas can infiltrate the lungs (known as Granulomatous-Lymphocytic Interstitial Lung Disease or GLILD), the liver (causing hepatomegaly and portal hypertension), and the spleen. Generalized lymphadenopathy and splenomegaly are present on physical examination in up to one-third of patients, reflecting benign but extensive lymphoid proliferation.
5. Increased Malignancy Risk
Patients with CVID carry a significantly elevated baseline risk for developing malignancies. There is a remarkably high incidence of gastric carcinoma, heavily associated with a higher rate of Helicobacter pylori infections and achlorhydria. Furthermore, the risk of developing non-Hodgkin lymphoma is increased up to 30-fold compared to the general population, typically emerging during adulthood.
4
Diagnosis & Workup
2 min•313 words
A definitive diagnosis of Common Variable Immunodeficiency requires fulfilling a strict, internationally recognized consensus criterion while carefully excluding alternative causes of hypogammaglobulinemia.
Quantitative Serum Immunoglobulin Quantification: The initial step is checking serum immunoglobulin levels. Diagnostic criteria require a marked reduction in serum IgG (typically below 400 mg/dL or at least two standard deviations below the age-matched mean), paired with a significant reduction in serum IgA, IgM, or both. These tests must be repeated and verified at least twice.
Evaluation of Functional Antibody Response: Demonstrating low antibody numbers is insufficient; the clinician must document a functional failure of antibody production. Evaluate the patient's specific IgG antibody titers against both protein antigens (e.g., Tetanus toxoid or Diphtheria vaccine) and polysaccharide antigens (e.g., Pneumococcal vaccine). If baseline titers are low, administer the standard vaccine challenge and re-evaluate titers 4 to 6 weeks later; a failure to mount a protective, multi-fold rise in IgG confirms a functional deficit.
Flow Cytometry Immunophenotyping: Perform a peripheral blood flow cytometry assessment to map lymphocyte subsets. This workup tracks the total counts of B-cells (CD19+), T-cells (CD3+, CD4+, CD8+), and Natural Killer cells. In CVID, total CD19+ B-cell percentages are usually normal or mildly decreased, but specialized flow cytometry will demonstrate a severe reduction in switched memory B-cells (CD19+CD27+IgD-IgM-).
Exclusion of Secondary Hypogammaglobulinemia: Clinicians must rigorously rule out alternative causes of low immunoglobulins before making a diagnosis of CVID. This includes checking for protein-losing enteropathies, nephrotic syndrome (via a urinalysis and spot protein-to-creatinine ratio), drug-induced hypogammaglobulinemia (e.g., due to Rituximab, glucocorticoids, or anti-epileptic agents like Phenytoin), and hematological malignancies such as Chronic Lymphocytic Leukemia or Multiple Myeloma via a serum protein electrophoresis.
Age Threshold: A formal diagnosis of CVID should generally not be established before the age of 4 years to allow for the complete physiological maturation of the child's humoral immune system and the natural clearance of maternal transplacental IgG.
Quantitative Serum Immunoglobulin Quantification: The initial step is checking serum immunoglobulin levels. Diagnostic criteria require a marked reduction in serum IgG (typically below 400 mg/dL or at least two standard deviations below the age-matched mean), paired with a significant reduction in serum IgA, IgM, or both. These tests must be repeated and verified at least twice.
Evaluation of Functional Antibody Response: Demonstrating low antibody numbers is insufficient; the clinician must document a functional failure of antibody production. Evaluate the patient's specific IgG antibody titers against both protein antigens (e.g., Tetanus toxoid or Diphtheria vaccine) and polysaccharide antigens (e.g., Pneumococcal vaccine). If baseline titers are low, administer the standard vaccine challenge and re-evaluate titers 4 to 6 weeks later; a failure to mount a protective, multi-fold rise in IgG confirms a functional deficit.
Flow Cytometry Immunophenotyping: Perform a peripheral blood flow cytometry assessment to map lymphocyte subsets. This workup tracks the total counts of B-cells (CD19+), T-cells (CD3+, CD4+, CD8+), and Natural Killer cells. In CVID, total CD19+ B-cell percentages are usually normal or mildly decreased, but specialized flow cytometry will demonstrate a severe reduction in switched memory B-cells (CD19+CD27+IgD-IgM-).
Exclusion of Secondary Hypogammaglobulinemia: Clinicians must rigorously rule out alternative causes of low immunoglobulins before making a diagnosis of CVID. This includes checking for protein-losing enteropathies, nephrotic syndrome (via a urinalysis and spot protein-to-creatinine ratio), drug-induced hypogammaglobulinemia (e.g., due to Rituximab, glucocorticoids, or anti-epileptic agents like Phenytoin), and hematological malignancies such as Chronic Lymphocytic Leukemia or Multiple Myeloma via a serum protein electrophoresis.
Age Threshold: A formal diagnosis of CVID should generally not be established before the age of 4 years to allow for the complete physiological maturation of the child's humoral immune system and the natural clearance of maternal transplacental IgG.
5
Management
3 min•407 words
The therapeutic management of Common Variable Immunodeficiency focuses on two main strategies: aggressive lifelong immunoglobulin replacement to prevent infections, and targeted immunosuppression to control the complications of immune dysregulation.
1. Immunoglobulin Replacement Therapy (IgRT)
Immunoglobulin replacement is the foundational, life-saving intervention for CVID. It provides immediate, passive humoral immunity using pooled IgG extracted from thousands of healthy blood donors. IgRT can be administered via two primary routes:
Intravenous Immunoglobulin (IVIG): Infused in a clinical setting once every 3 to 4 weeks, typically at a starting dose of 400 to 600 mg/kg. It produces rapid, high peak serum IgG levels but can be associated with systemic side effects like headaches, flushing, or aseptic meningitis.
Subcutaneous Immunoglobulin (SCIG): Self-administered by the patient or caregiver at home once a week or every other week using a small portable infusion pump. SCIG provides highly stable, steady-state trough IgG levels with fewer systemic adverse effects, making it an excellent choice for patients with poor venous access or those who experience systemic reactions to IVIG.
The primary therapeutic goal is to individualize the dose to keep the trough serum IgG level well within the normal range (ideally above 700 to 800 mg/dL) and, more importantly, to completely eliminate recurrent sino-pulmonary infections.
2. Prophylactic and Aggressive Antibiotic Protocols
Despite adequate IgRT, some patients continue to experience structural airway infections. Acute bacterial infections must be treated early and aggressively with prolonged, full-dose courses of broad-spectrum oral or intravenous antibiotics. In patients who develop established bronchiectasis and suffer from frequent pulmonary exacerbations, long-term antibiotic prophylaxis—such as oral Azithromycin administered three times per week—may be implemented to reduce the bacterial load and limit structural lung degradation.
3. Management of Autoimmune and Inflammatory Complications
Treating the non-infectious complications of CVID presents a major therapeutic paradox, as it requires administering immunosuppressive therapies to a patient who is already profoundly immunodeficient.
Cytopenias and Autoimmunity: High-dose systemic Corticosteroids (e.g., Prednisolone) remain the first-line therapy for acute episodes of CVID-associated ITP or AIHA. For corticosteroid-resistant or refractory cases, the monoclonal anti-CD20 antibody Rituximab is highly effective at eliminating the aberrant autoantibody-producing B-cell clones, though it will further depress endogenous antibody production.
Granulomatous Disease (GLILD): Requires combination immunosuppressive therapy, frequently pairing Corticosteroids with steroid-sparing agents such as Azathioprine or Mycophenolate Mofetil to halt progressive pulmonary fibrosis.
Enteropathy: Mild cases may respond to oral budesonide, while severe, refractory enteropathy requires systemic immunosuppression or biological agents like anti-TNF alpha therapy, managed under close specialist surveillance.
1. Immunoglobulin Replacement Therapy (IgRT)
Immunoglobulin replacement is the foundational, life-saving intervention for CVID. It provides immediate, passive humoral immunity using pooled IgG extracted from thousands of healthy blood donors. IgRT can be administered via two primary routes:
Intravenous Immunoglobulin (IVIG): Infused in a clinical setting once every 3 to 4 weeks, typically at a starting dose of 400 to 600 mg/kg. It produces rapid, high peak serum IgG levels but can be associated with systemic side effects like headaches, flushing, or aseptic meningitis.
Subcutaneous Immunoglobulin (SCIG): Self-administered by the patient or caregiver at home once a week or every other week using a small portable infusion pump. SCIG provides highly stable, steady-state trough IgG levels with fewer systemic adverse effects, making it an excellent choice for patients with poor venous access or those who experience systemic reactions to IVIG.
The primary therapeutic goal is to individualize the dose to keep the trough serum IgG level well within the normal range (ideally above 700 to 800 mg/dL) and, more importantly, to completely eliminate recurrent sino-pulmonary infections.
2. Prophylactic and Aggressive Antibiotic Protocols
Despite adequate IgRT, some patients continue to experience structural airway infections. Acute bacterial infections must be treated early and aggressively with prolonged, full-dose courses of broad-spectrum oral or intravenous antibiotics. In patients who develop established bronchiectasis and suffer from frequent pulmonary exacerbations, long-term antibiotic prophylaxis—such as oral Azithromycin administered three times per week—may be implemented to reduce the bacterial load and limit structural lung degradation.
3. Management of Autoimmune and Inflammatory Complications
Treating the non-infectious complications of CVID presents a major therapeutic paradox, as it requires administering immunosuppressive therapies to a patient who is already profoundly immunodeficient.
Cytopenias and Autoimmunity: High-dose systemic Corticosteroids (e.g., Prednisolone) remain the first-line therapy for acute episodes of CVID-associated ITP or AIHA. For corticosteroid-resistant or refractory cases, the monoclonal anti-CD20 antibody Rituximab is highly effective at eliminating the aberrant autoantibody-producing B-cell clones, though it will further depress endogenous antibody production.
Granulomatous Disease (GLILD): Requires combination immunosuppressive therapy, frequently pairing Corticosteroids with steroid-sparing agents such as Azathioprine or Mycophenolate Mofetil to halt progressive pulmonary fibrosis.
Enteropathy: Mild cases may respond to oral budesonide, while severe, refractory enteropathy requires systemic immunosuppression or biological agents like anti-TNF alpha therapy, managed under close specialist surveillance.
6
Key Pearls & Takeaways
1 min•200 words
Look for the Variable Age: Do not rule out a primary immunodeficiency just because the patient is an adult. CVID is most frequently diagnosed in patients between 20 and 40 years of age who present with a new history of recurrent pneumonias.
The Evans Syndrome Red Flag: Any adult or adolescent presenting with unexplained, concurrent, or sequential immune thrombocytopenia and autoimmune hemolytic anemia (Evans Syndrome) must immediately undergo quantitative immunoglobulin testing to rule out underlying CVID.
Vaccine Challenge is Mandatory: A low IgG number alone is not enough to diagnose CVID. You must document a functional failure of the immune system by demonstrating the patient's inability to produce specific IgG antibodies following a vaccine challenge.
Absence of Plasma Cells: If a patient undergoes a duodenal biopsy for suspected celiac-like chronic diarrhea, the complete and total absence of plasma cells in the lamina propria is a classic diagnostic clue pointing directly to CVID.
Transfuse the Dose, Track the Trough: Immunoglobulin replacement therapy is a lifelong commitment. Trough IgG levels must be monitored regularly and kept above 700-800 mg/dL, but the true marker of adequate dosing is a clinically stable patient who is completely free of recurrent sinus and lung infections.
References
The Evans Syndrome Red Flag: Any adult or adolescent presenting with unexplained, concurrent, or sequential immune thrombocytopenia and autoimmune hemolytic anemia (Evans Syndrome) must immediately undergo quantitative immunoglobulin testing to rule out underlying CVID.
Vaccine Challenge is Mandatory: A low IgG number alone is not enough to diagnose CVID. You must document a functional failure of the immune system by demonstrating the patient's inability to produce specific IgG antibodies following a vaccine challenge.
Absence of Plasma Cells: If a patient undergoes a duodenal biopsy for suspected celiac-like chronic diarrhea, the complete and total absence of plasma cells in the lamina propria is a classic diagnostic clue pointing directly to CVID.
Transfuse the Dose, Track the Trough: Immunoglobulin replacement therapy is a lifelong commitment. Trough IgG levels must be monitored regularly and kept above 700-800 mg/dL, but the true marker of adequate dosing is a clinically stable patient who is completely free of recurrent sinus and lung infections.
References
0/6